大阪大学は2019年6月27日、キンギョの全ゲノム配列の解読に成功したと発表した。約1400万年前にキンギョの祖先種で全ゲノム重複が起こったことや、倍加した遺伝子の一部が淘汰、進化を遂げる様子を解明した。国立遺伝学研究所、愛知県水産試験場内水面漁業研究所、アメリカ国立衛生研究所との共同研究による成果だ。
今回の研究では、1万~4万塩基を一気に解析できる新型ロングリード次世代DNAシーケンサーを使用。旧型シーケンサーの100倍以上の長さのDNA配列を読み進められるため、配列不明の部分を減らし、正確な遺伝子地図を作成できる。
また、解析するゲノム構造をシンプルにするために、母親由来の染色体のみからなる雌性発生キンギョを作製した。キンギョ種は、野生のフナに近い体形を持つワキンという品種を用いた。
その結果、全ゲノム配列の解析が可能となり、約1400万年前に全遺伝子が倍加(全ゲノム重複)したことが分かった。さらに、倍加した遺伝子の12%が淘汰によって消失したこと、重複遺伝子の約30%が新たに臓器で発現するなど進化を遂げていることが明らかになった。遺伝子が失われる速度は、8000万年前に全ゲノム重複が起こったサケと比べて1.7倍速かった。
キンギョは形態が多様で、ヒトを含む脊椎動物の形や色を作るメカニズム解明に役立つと考えられている。本研究の成果は、ヒトの病気の原因解明や診断・治療法の確立にもつながる可能性がある。研究グループは今後、デメキンやランチュウを含むさまざまなキンギョ品種のゲノム解析を行うことで、脊椎動物の体を作る遺伝子の探索を検討している。
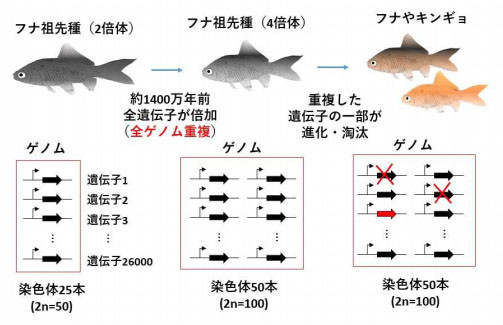
キンギョとフナの祖先種での全ゲノム重複と進化 出典:大阪大学
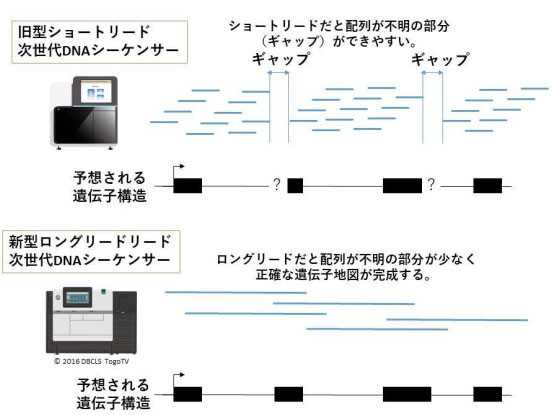
ロングリード次世代DNAシーケンサーによるゲノム解読 出典:大阪大学
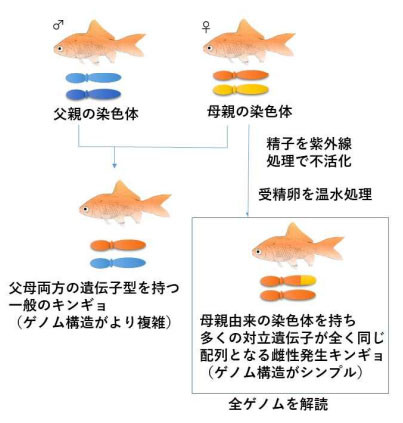
雌性発生キンギョの作出と染色体 出典:大阪大学
